

Briefings in Bioinformatics: 电子科技大学邹权组研发自动构建基于肠道微生物平衡的疾病预测模型及微生物生物标志物发掘平台


DisBalance: a platform to automatically build balance-based disease prediction models and discover microbial biomarkers from microbiome data
Article,2021-04-08
Briefings in Bioinformatics, [IF 9.101]
DOI:https://doi.org/10.1093/bib/bbab094
第一作者:Fenglong Yang (杨凤龙)
通讯作者:Quan Zou (邹权)
主要单位:
电子科技大学基础与前沿研究院(Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, Shahe Campus:No.4, Section 2, North Jianshe Road, 610054, China)
通过高通量测序以及相关数据库注释后获得的微生物组丰度数据包括功能分类单元(代谢途径,蛋白家族等)或物种分类单元(比科属种OTU等)等谱矩阵。谱矩阵描述了样本中功能或所属物种的丰度信息。谱矩阵常常是成分数据(compositional data),各成分(component)间相加总和为常数,即具有“定和限制”的特性,其中一个成分的减少,必然伴随其他一个或多个成分的增加。微生物组数据的组成性问题常常被忽视,进而导致误导性的发现、相互矛盾的解释和不可复现的分析结果。正确使用相应的数学理论分析微生物组数据对挖掘与人类健康疾病真正相关联的生物学标记和特征具有重要的意义。
应用监督学习可以从微生物组数据中识别可预测人类疾病表型的微生物特征(也称为生物标志物)。这些微生物生物标志物可用于个人健康监测,疾病预防治疗等。然而,如何对高维且具有组成性特点的微生物组数据进行正确的特征提取是决定疾病分类模型性能和可靠性的关键步骤。Quinn和Erb 评估了数据变换(包括原始的比例数据和5种“对数比”变换方法)对高维生物标志物成分数据的分类准确性的影响。对数比变换包括中心对数比变换(CLR)和4种基于Balance变换的方法(DBA、RDA、ARA、PBA)。结果表明,CLR 和4种balance变换方法在大多数分类任务中表现相当出色,均优于基于相对丰度特征的分类结果。
解决成分数据问题的研究通常应用加法对数比(the additive log-ratio,ALR)、中心化对数比(centred log-ratio,CLR)、等距测对数比(isometric log-ratio,ILR)来解决在微生物组数据的问题。成分数据的样本空间是一个受约束的单形空间(simplex),在同维度的单形空间和欧式空间之间找到等距映射(样本相对距离保持不变)对于多种多元统计工具在成分数据上的应用至关重要。由于ALR变换不是等距变换,CLR变换也没有用单形空间的一组正交基来表示,所以这个问题只能通过ILR变换来解决。
ILR变换表示为单形空间中的标准正交基,在成分数据分析中最经常被推荐使用。ILR变换方式的不同不会导致随后分析结果的不一致。ILR变换常用顺序二进制划分方法(sequential binary partition,SBP)来表示。然而,基于任意ILR变换的分析结果通常难以解释,为了克服这一困难,Egozcue和Pawlowsky-Glahn等人提出了“Balance”变换(一种特殊的ILR变换)。Balance变换的目的不是寻找与目标表型相关的单一特征,而是以可解释的方式寻找重要的特征比率(如细菌丰度比率)。虽然一些临床微生物组的研究中强调了将Balance作为生物标志物的重要性,但只有少数研究将Balance作为疾病预测等监督学习的输入特征。“selbal”方法首次成功地将Balance思想应用于基于微生物组数据的机器学习分类任务中,该方法旨在筛选出由多个组分构成的分类表现良好的唯一一个Balance特征。为了进一步提高当前基于Balance特征提取方法的分类性能与可解释性,Quinn和Erb应用了计算高效的判别平衡分析(discriminative balance analysis,DBA-distal)方法来筛选由2个或3个组分构成的,具有优良分类判别能力的Balances特征。针对基于微生物组数据的表型分类问题,DBA-distal方法在可解释性、运算时间和分类准确性方面的表现均优于selbal方法。
我们前期发表的工作(图1,GutBalance),成功应用DBA-distal方法对GMrepo数据库中数十种人类肠道微生物组有关的疾病数据集进行了深入分析,我们构建了基于distal balances特征的数十种疾病的预测模型数据库(GutBalance),并开发了在线疾病预测平台以及疾病微生物标志物数据库GBDAD,系统解决了基于肠道微生物组数据进行疾病预测和生物标志物发掘的问题。

图1 GutBalance,基于Balance特征的疾病预测模型及生物标志物数据库(http://lab.malab.cn/soft/GutBalance)。
由于基于肠道微生物组的人类疾病分类预测研究普遍缺乏对成分数据特点的考虑,我们强调“平衡-疾病”关联的概念,而不是传统的“微生物-疾病”关联,并研发了一站式基于成分数据的疾病预测建模综合分析平台(图2,DisBalance)。该平台集成并简化了基于微生物组数据的疾病二分类建模、风险预测和疾病相关生物标志物发现的分析流程,可以快速方便地构建新的疾病预测模型,辅助与人类肠道菌群相关的疾病诊断和预防,促进模型驱动和知识驱动的机制发现。
图2 DisBalance,基于微生物组数据的Balance变换及疾病预测建模分析平台(http://lab.malab.cn/soft/DisBalance)。
DisBalance数据分析平台介绍
分析流程
DisBalance平台采用DBA-distal方法选择一组具有疾病判别能力的Balances作为正则化逻辑回归(LR)建模的输入特征,平台内置了从MicroPhenoDB数据库中提取的2111个疾病-微生物关联证据,提供多种挖掘微生物标志物的策略,分析流程见图3。DisBalance在线平台,对于微生物-疾病关联研究中疾病预测建模、风险预测和微生物标志物发现等都具有重要的意义和临床应用价值。
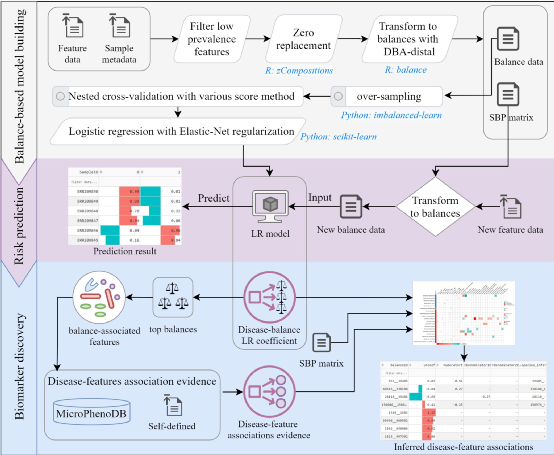
图3 DisBalance平台由三个功能模块组成:1. Logistic regression(LR)模型构建,2.疾病风险预测 3.生物标志物发现。执行LR建模流程,只需输入两个文件:表示特征丰度信息的谱矩阵文件和包含样本表型分类信息的样本元数据文件。首先,过滤低组内覆盖度的特征(默认20%),采用多重简单置换的方法替换零值;然后采用DBA-distal方法生成SBP矩阵,从该矩阵中选择组成末端Balances的物种,并通过ILR(等距对数比)变换获得Balance谱数据(Balance-样本矩阵);应用SMOTE采样技术处理类不平衡的数据集后,输入正则化的LR模型,利用嵌套交叉验证的方法进行训练得到优化后的疾病预测模型。根据构建好的疾病预测模型,一方面,可以提交新的物种丰度数据来预测新样本的患病风险;另一方面,有四种策略可以帮助发现微生物生物标志物:1)通过筛选LR系数绝对值最大的top balances作为候选生物标志物,2)top balances相关的微生物也可以作为候选生物标志物,3)top balances相关的微生物可以通过已发表的疾病关联证据(如MicroPhenoDB微生物-疾病关联数据库)进行佐证,4)新的微生物-疾病关联可以通过结合top balances和实证的疾病相关微生物来进行推断,DisBalance平台内置MicroPhenoDB数据库,也支持上传自定义的微生物-疾病关联证据。
DisBalance平台的Web应用程序使用基于Python语言的 Django前端框架和Plotly Dash框架实现。该平台部署在阿里巴巴弹性计算云服务器上。由于服务器的存储有限,用户提交的数据和平台生成的数据需要及时下载,所有数据将在任务完成后24小时后自动删除。
平台输出的可视化结果
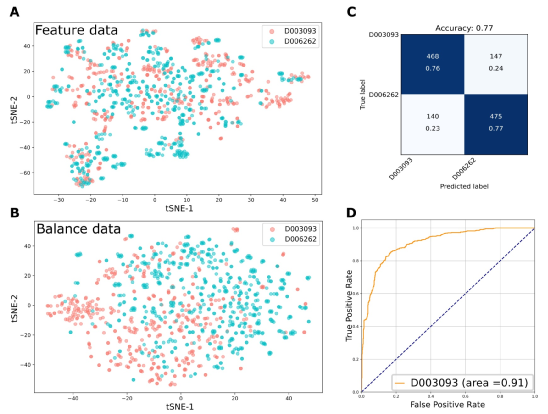
图4以UC(D003093)疾病模型为例,展示DisBalance建模结果的可视化。包括基于物种丰度(A)和Balance(B)的tSNE样本分布图、混淆矩阵热图(C)以及模型评估ROC曲线(D)。很明显,基于Balance特征可以更好地区分疾病与健康样本。
图4展示了平台分析结果模型的可视化,其他分析结果见服务器Demo部分介绍(http://lab.malab.cn/soft/DisBalance)。
疾病-微生物关联推断
疾病预测模型筛选出来的top balances特征(按LR系数绝对值大小排序的top balances)和与top balances相关的微生物都可以作为候选的疾病标志物。Top balance相关的微生物与疾病的关联可以从MicroPhenoDB数据库中获得佐证。新的疾病-微生物关联可以通过结合疾病-Balance关联和实证的疾病-微生物关联来进行推断(图5B)。从图5A中,我们可以很方便的找到balances对应的微生物是否与MicroPhenoDB数据库中的相关疾病有关。
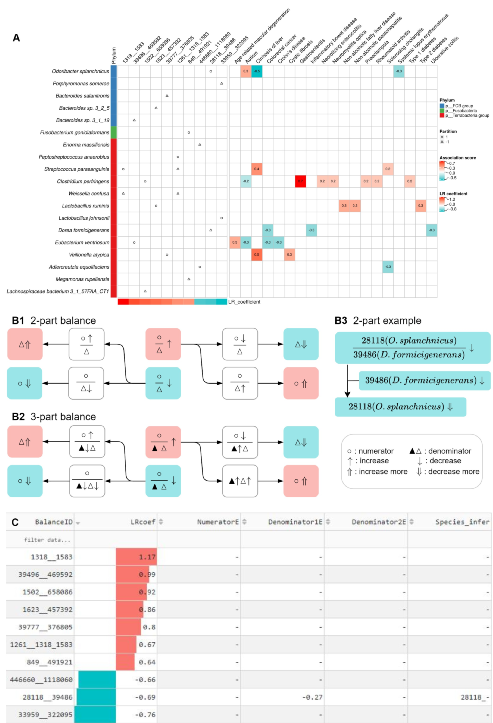
图5疾病-微生物关联推断方法(以UC数据集为例)。A. Top 10 balances的物种划分情况以及 MicroPhenoDB中与top 10 balance相关的微生物与疾病关联的证据,红色代表正相关,蓝色代表负相关。Balance中分子分母处的物种分别用圆圈和三角形表示。B. 结合疾病-Balance关联和实证的疾病-微生物关联推断新的疾病-微生物关联。对于与UC疾病正(红色)或负(蓝色)相关的2-part(B1)或3-part(B2)的balances,各自有四种情况可以唯一推断出新的疾病-微生物关联。以UC的2-part balance(28118_39486)为例(B3),模型训练结果显示该balance与UC负相关,balance中的分母物种Dorea formicigenerans(NCBI id: 39486)被证明与UC负相关,那么推断出分子物种Odoribacter splanchnicus(NCBI id:28118)与UC负相关。C. DisBalance平台的推断结果为在线交互表格,包含了top balances信息、与疾病相关的LR系数、MicroPhenoDB中实证的疾病-微生物关联信息以及新的疾病-微生物关联推断结果。
为了简化推论过程,我们设计了一个功能模块,可以自动从top balances和实证的疾病-微生物关联中推断出新的疾病-微生物关联。DisBalance默认使用MicroPhenoDB中的疾病-微生物关联证据,也支持用户上传自定义的疾病-微生物关联证据进行推论。推断结果可以通过输出界面进行交互式探索(图5C)。
本研究开发了针对基于微生物组数据进行疾病分类研究的一站式在线分析平台DisBalance。该平台整合了微生物组与疾病研究中涉及的疾病模型构建、疾病风险预测和疾病微生物标志物挖掘等分析内容,可以快速方便地对提交的数据进行模型构建和疾病风险预测。为了促进模型驱动的知识发现,DisBalance 提供了四种策略来帮助自动挖掘疾病相关的微生物标志物。UC实例数据的分析证明了流程的可靠性及使用方法。
我们强调Balance-疾病关联研究框架,并提供了端到端的相应研究方案与在线软件工具,系统解决了微生物组数据组成性问题对分析结果可靠性的影响。基于Balance研究框架,我们进一步创新性地提出了基于Balance建模结果的疾病-微生物关联推断方法和大规模数据挖掘结果,为疾病-微生物关联的实证研究与临床实践提供了指导。
影响疾病预测模型可信度的因素都会影响微生物-疾病关联的推断。这些因素包括:疾病健康样品的代表性、高通量测序方法及物种注释方法的选择、零值替换以及微生物-疾病关联证据的可靠性等。
1. 微生物稀疏矩阵大量零值的替换。目前有不同的方法可以替换稀疏矩阵的零值,本研究使用了简单的多重置换方法。零值的处理方式将直接影响基于对数比的特征选择结果,进而影响训练模型的准确度,而如何最好地替换零值仍然是一个待解决的问题。
2. 预测模型及模型推测的balance-疾病关联的可靠性。我们选择正则化的LR而不是其他机器学习算法来解决基于肠道微生物数据的疾病分类预测问题。LR算法的优势在于两个方面。首先,LR 模型具有高度可解释性,LR变量权重系数可直接用作衡量特征重要性的指标以及与表型的相关性。其次,通过应用弹性网正则化以及使用嵌套交叉验证的方法来训练LR模型可以有效规避模型对数据的过拟合问题。
3. 微生物-疾病关联证据的可靠性。虽然本研究使用的MicroPhenoDB是与人类微生物-疾病关联证据收集最新的数据库,但规模仍然有限。构建全面的微生物-疾病关联的知识数据库有望加速人类微生物-疾病关联数据的整合。新的微生物-疾病关联研究的不断推进也将促进GBDAD数据库中与疾病相关的balances或微生物致病机理的揭示。
目前,DisBalance是解决基于微生物组数据进行疾病二分类建模的最佳解决方案。基于DBA-distal的对数比变换方法尚未完全解决微生物组数据的计数(count data)问题,因此建议将DisBalance应用于样本量较大的数据集。除了我们在本研究中关注的微生物物种特征外,DisBalance还可以用于分析其他类型的成分数据,如基因、代谢物和mRNA等。
参考文献
Fenglong Yang, Quan Zou, DisBalance: a platform to automatically build balance-based disease prediction models and discover microbial biomarkers from microbiome data, Briefings in Bioinformatics, 2021, Doi: 10.1093/bib/bbab094.
第一作者简介

第一作者:杨凤龙,电子科技大学基础与前沿研究院博士后(已出站),主要关注肠道微生物组与人类健康,人工智能算法等。目前以第一作者在Briefings in Bioinformatics,Database(Oxford)等期刊发表多篇论文,开发了多个微生物组数据分析流程及web服务。目前担任计算基因组学(Computational Genomics)Frontiers in Genetics, Frontiers in Bioengineering and Biotechnology, Frontiers in Plant Science 等杂志的评审编辑,以及Journal of Electronic Science and Technology等杂志审稿人。
通讯作者简介

邹权,电子科技大学基础与前沿研究院教授,博士研究生导师,IEEE高级会员,ACM高级会员,CCF杰出会员,科睿唯安全球高被引学者。2009年获哈尔滨工业大学博士学位,研究领域是生物信息学,机器学习和并行计算。先后主持国家自然科学基金优秀青年基金、国家自然科学基金面上项目和四川省杰出青年科技人才等项目,研究成发表在生物信息主流刊物PLoS Computational Biology, RNA, Bioinformatics, Briefings in Bioinformatics等。
邹老师课题组目前常年招聘博士后,具体请参考如下链接:http://lab.malab.cn/~zq/recruit/postdoc2020.html。
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”